First-principles modeling are essential and important to predict material properties. Modelling analysis are also necessary to understand the intricate details about material properties that are not accessible with experimental methods. Moreover, materials have their unique ways to respond to external stimuli such as application of pressure, temperature, electric, optical, and magnetic fields among others.
HIRG employs a combination of density functional theory (DFT) and molecular dynamics (MD) simulation methods to understand the structural, electronic, and thermal properties of materials. These simulations help us not only explain various experimental results that we obtain, but also to predict new properties. At the same time, the group also uses standard device simulation techniques to predict device properties.
|
 |
Publications:
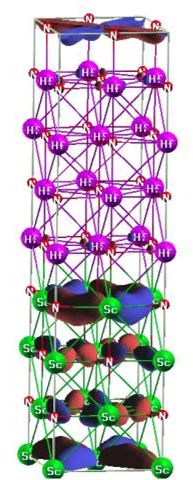
1. B. Saha, T. D. Sands and U. V. Waghmare, "Electronic structure, vibrational spectra and thermal properties of HfN/ScN metal/semiconductor superlattices: A first-principles Study." J. Phys.: Cond. Matt., 24 415303, (2012). 
2. B. Saha, T. D. Sands and U. V. Waghmare, "Electronic structure, vibrational spectrum, and thermal properties of yttrium nitride (YN): A first-principles study." J. Appl. Phys. 109, 083717 (2011).
3. B. Saha, T. D. Sands and U. V. Waghmare, "First-principles analysis of thermoelectric ZrN/ScN metal/semiconductor superlattices", J. Appl. Phys. 109, 073720 (2011).
4. B. Saha, J. Acharya, T. D. Sands and U. V. Waghmare, "Electronic structures, phonons and thermal properties of ScN, ZrN and HfN: A first-principles Study", J. Appl. Phys. 107, 033715 (2010).
|








